

 Processing
Processing

Traditional methods of site-directed mutagenesis
In vitro site-directed mutagenesis is a core technique in functional genomics studies. Until recently, labs performing site-directed mutagenesis relied primarily on PCR and standard cloning methods. When PCR is used, the primers are designed to include the desired change, which might be a base substitution (Figure 1), addition (Figure 2), or deletion (Figure 3). During amplification, the mutation is incorporated into the amplicon, altering the original sequence.
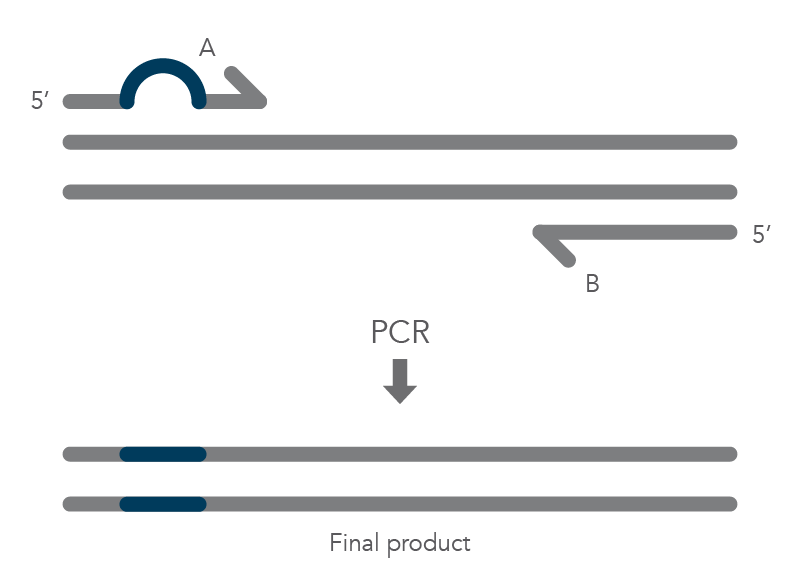
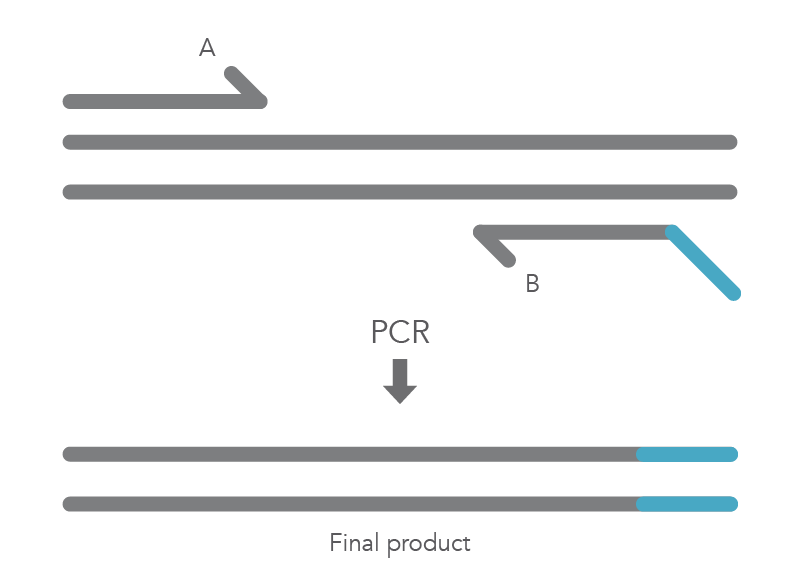
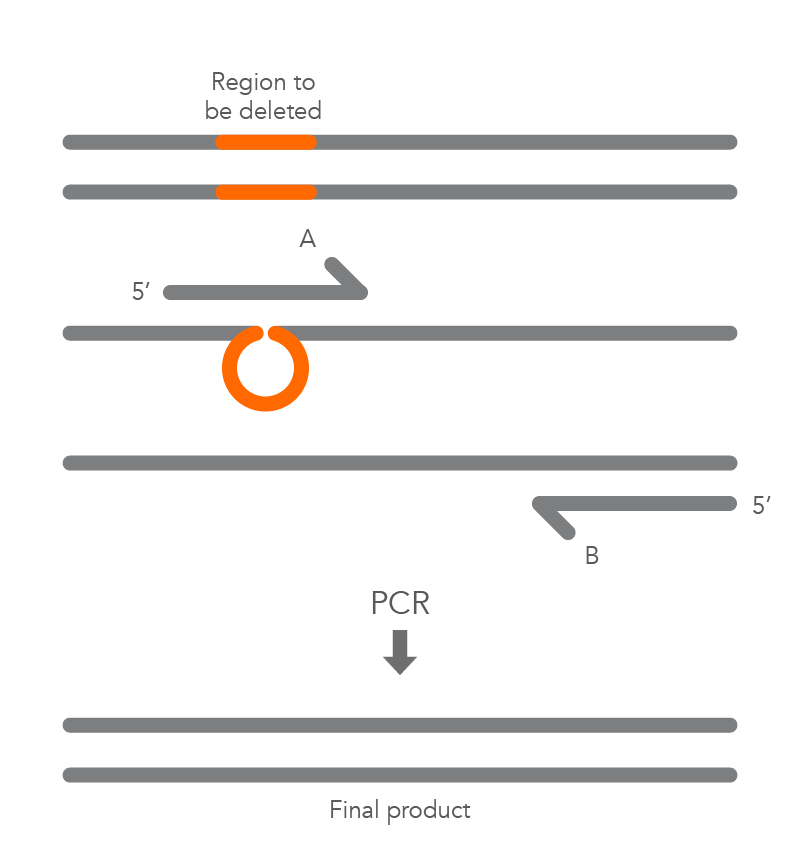
Introducing mutations with long primers through PCR works for many applications, but it also has limitations. One of these is that the size of the mutation is constrained by the maximum length of the DNA oligonucleotide used. And as oligo length increases, the percentage of full-length product decreases, resulting in less efficient PCR and a lower percentage of the desired construct.
To address these issues, IDT offers high quality Ultramer ™ Oligonucleotides up to 200 bases. These long oligos are synthesized using a proprietary coupling process, enabling IDT to provide the highest percentage of full-length product in the industry.
Ultramer ™ Oligonucleotides are the best option for long mutagenesis oligos due to their quality, and high coupling efficiency. However, some of the available 200 base sequence must be complementary to the original sequence, which still limits the maximum possible sequence insertion length to <200 bases. Furthermore, if you want to alter the sequence at more than one site (e.g., changing several codons at one site, and adding a protein tag to the resulting mutant), you will need to perform multiple rounds of mutagenesis, and the cost of reagents and time will rise accordingly. Other challenges for use of long oligos include that they do not perform efficiently in applications involving high GC content or secondary structure, and very long oligos are more expensive per base than recently developed alternatives.
Synthetic gene fragments reduce the work
Arguably, the most significant improvement to mutagenesis methods is the commercial availability of long, synthetic, double-stranded (ds), custom DNA fragments. IDT provides them as eBlocks™, gBlocks™, and gBlocks HiFi Gene Fragments. Up to 3 kb dsDNAs can be obtained with the desired mutations designed directly into the sequences. They are compatible with a variety of cloning methods from PCR and restriction cloning to seamless cloning methods. By using a seamless cloning method, researchers can eliminate some of the time-consuming steps needed to produce both a wild-type sequence and any derivative variants. Now, for a reasonable cost, researchers can design all the requisite sequences for their experiments, order them online, and receive them ready for direct use or cloning. Moreover, final constructs can be easily generated even when a physical starting template is not available.
Seamless cloning methods for mutagenesis
Seamless cloning (which includes isothermal assembly methods) is an easy way to make seamless changes to gene sequences. Isothermal mechanisms assemble pieces of linearized DNA—typically, a plasmid and one or more inserts—with overlapping homologous ends by first modifying the DNA and then joining the fragments [2]. Typically, the fragments are mixed together in the reaction, and overhanging ends are created by an enzyme with endonuclease activity. The resulting “sticky” ends then anneal to the complementary fragments, which determines the precise position and directionality of each piece in the finished construct. A polymerase then fills in the gaps, and a ligase seals the nicks (Figure 4).

Figure 4. An overview of the seamless cloning method. This method incorporates cloning of more than one DNA fragment into a vector. Although this method does not require restriction enzyme digestion, the vector and the DNA fragments require insertion of a 15–40 base pair sequence overhang (depicted as orange and blue in the figure). Instead, the DNA fragments and an enzyme master mix containing 5’ exonuclease, DNA polymerase, and DNA ligase are combined in a single-tube reaction. After an incubation period, the assembled construct is transformed into a host organism then plated on a solid agar growth medium.
For isothermal assembly, the plasmid can be linearized by restriction digest or PCR, giving you the freedom to insert your sequence at any location regardless of the presence of restriction sites. However, a benefit of using PCR to linearize you vector is that any circularized plasmids can be removed by DpnI digest, reducing the presence of empty vectors when screening clones for finished sequences.
Double-stranded variable libraries
Often labs need more than one version of a particular construct. When only a few variants are required, it can save significant time and resources to order all of the variants as standard eBlocks or gBlocks Gene Fragments. However, for mutagenesis applications where you need to test many different amino acid combinations (e.g., antibody research, binding affinity studies, and enzyme kinetic studies) variable bases can be included in the gBlocks Gene Fragment sequence to create a diverse pool of sequences (gBlocks Gene Fragments Libraries).
Variable bases can be used to study structural DNA elements like promoters and enhancers and are commonly used to create NNK libraries of amino acid codon variants. In NNK libraries, N represents DNA bases A, G, C, T, and K represents bases G and T. By using the NNK variable string in your sequence design, you can make a random library with 32 of the 64 codons, and simultaneously eliminate 2 of the 3 stop codons. The NNK codons cover all of the standard amino acids; limiting to 32 codons makes it easier to screen your finished library. Until recently, the creation of mixed base libraries had to be accomplished by synthesis and then pooling of separate oligonucleotides. However, unlike other synthetic dsDNA products, gBlocks Gene Fragments can also be designed to contain regions of variable bases.
Currently, the online ordering tool allows you to incorporate up to 18 sequential N or K bases, generating libraries with >1 billion variants. However, other custom designs that are not supported by the automated, online ordering system may still be feasible by contacting the experts using the IDT Help Request Form.
When does it make sense to use oligonucleotides versus a gene fragment?
Consider which dsDNA option makes the most sense for your mutagenesis application. Table 1 offers some examples of how oligonucleotides or gene fragments can be used in common mutagenesis applications.
| I want to: | Oligonucleotides | IDT Gene Fragments |
|---|---|---|
| Create a unique sequence and a variant of that sequence. | Oligos can be used to create genes de novo, however the processes are complex, time consuming and do not always lead to success. | Synthetic gBlocks Gene Fragments are ideal for this application. Both the wild type and mutant versions can be ordered as complete constructs (up to 3 kb) and cloned simultaneously, saving time and money. Longer constructs can be assembled from multiple fragments. |
| Make a single mutation in a sequence cloned in a plasmid. | Standard oligos work well for making small changes and Ultramer Oligos can be used to make larger changes. However, the cost of long oligos can often exceed the cost of a gBlocks Gene Fragment. Long PCR methods may also require more optimization than alternative gene assembly approaches such as isothermal assembly. | IDT Gene Fragments are a good option, using a seamless cloning method gBlocks can be used to create changes up to 3 kb within an existing plasmid. Researchers should consider the cost of materials and the assembly method they prefer to use (i.e., PCR mutagenesis or gene assembly). |
| Make changes to multiple sites to a sequence cloned into a plasmid. | Using oligos, several steps will be needed to create the mutant, with screening at each stage. | IDT Gene Fragments are ideal for this application. Because their sequence is completely customizable, large segments of the sequence (up to 3 kb) can be replaced at once, saving both time and resources in the lab. |
| Create a library to screen every possible sequence combination at a specific site or sites within a sequence. | For simple variable base insertions, such as a single codon or a few nucleotides, oligonucleotide mutagenesis often works well. However, for more complex applications, the assembly and verification of evenness of the library can be difficult and time consuming. | gBlocks Gene Fragments can be ordered with N or K variable bases. Simple libraries can be ordered directly from the web, and our design group can assist more complex library designs. Custom QC, including next generation sequencing (NGS) to verify the distribution of variation in the library can also be arranged. |
When planning your mutagenesis experiments, consider the following 3 factors to help you determine the best protocol and reagents for your purposes:
- Appropriateness of the methods and materials for your needs
- Familiarity with mutagenesis methods
- The cost of generating the final sequences
When selecting appropriate methods and materials, the size and number of proposed changes are important factors. Using high-fidelity oligos and mutagenesis reagents instead of the dsDNA fragments described above is still a viable method when the gene of interest is already cloned, and the desired mutation is relatively short.
For example, high quality Ultramer Oligonucleotides of up to 200 bp can be used to make changes of 100 bp or more. However, the efficiency of the mutagenesis reaction will decrease as the size of the change increases, and the cost of an oligo may be greater than the double-stranded DNA fragment.
Researchers may also want to continue to use long, single–stranded oligos and PCR based mutagenesis if they have experience with this approach. The labs that this applies to understand long PCR parameters and reagents and generally have a good understanding of critical design parameters for oligos used in mutagenesis. Though, these labs may also want to look at the cost benefits of dsDNA for longer sequence changes, even when the procedure is feasible with long oligos. Cost is often the first consideration when designing mutagenesis experiments. However, it can be difficult to determine the real expense until after completing construct assembly and verification. Up front, it is easy to calculate the costs of oligos, dsDNA, and enzyme kits. The costs of time spent and repeating failed procedures cannot be determined at the outset and should be controlled as much as possible by good planning and assessment of available expert advice. IDT has a skilled technical support team that can quickly assist you with many aspects of mutagenesis and gene assembly, available via the IDT Help Request Form.
IDT provides both high quality custom oligonucleotides and dsDNA fragments to facilitate these methods. For additional information about site-directed mutagenesis, download the Mutagenesis Handbook. The handbook provides an overview of in vitro mutagenesis experiment applications, protocols, and troubleshooting.
To learn more, download, IDT’s DNA Cloning Guide to read about common cloning methods, important sequence design considerations, and other tips when conducting your cloning experiment.
