

 Processing
Processing

Cloning of double-stranded DNA (dsDNA) molecules into plasmid vectors is a commonly employed technique in molecular biology. The procedure is used for sequencing, building libraries of DNA molecules, expressing coding and non-coding RNA, and many other applications.
Blunt-end cloning
Blunt-end cloning involves ligating dsDNA into a plasmid where both the insert and linearized plasmid have no overhanging bases at their termini. It does not benefit from the hydrogen bond stabilization associated with the complementary overhanging bases used in cohesive end cloning, but the transient associations of the available
5’ phosphate and 3’ hydroxyl groups are sufficient to produce successful clones in the presence of T4 ligase [1]. An illustration of a basic blunt-end cloning experiment is shown in Figure 1.
As an easy and versatile method for cloning dsDNA into plasmid vectors, researchers can avoid the enzymatic digestion and subsequent purification needed for cohesive end cloning. In addition, the insert and vector have fewer sequence limitations compared to other methods as discussed below.
Blunt-end cloning considerations
There are several factors to consider when performing blunt-end cloning. Blunt-end cloning is not directional as ligation of fragments with phosphate groups on both ends can occur in either direction. In addition, blunt-end cloning efficiencies decrease due to re-ligation of empty vectors if the vector is not dephosphorylated prior to cloning. Due to these considerations, when using this method, you should check for colonies with insert by restriction digestion or PCR prior to sequence-verification.
Blunt-end cloning advantages
A major advantage of blunt-end cloning is that the desired insert does not require any restriction sites in the sequence. This makes blunt-end cloning versatile, simplifies planning, and minimizes unwanted, artificial sequence additions that might adversely affect some applications. Also, because the insert does not need to be prepared by restriction digestion, blunt-end cloning has the potential to be faster than other cloning methods.
Tips for successful blunt-end ligations:
Preparing the vector.
The vector can be prepared by digestion if the multiple cloning site (MCS) contains a recognition site for a restriction enzyme that produces blunt ends, such as EcoRV (Figure 1). Restrictions sites that generate sequence overhangs can also be used, followed by removal or filling of the overhangs to create blunt ends. However, this approach is not recommended since there is no good method for assessing the success of the blunting reaction, making it hard to troubleshoot unsuccessful reactions.
Alternatively, linearized plasmid can be prepared for blunt-end cloning by PCR amplification with a high-fidelity polymerase and using primers designed with their 5’ ends at the desired insertion site. The linearized plasmid product appears as a distinct band on an agarose gel, compared to a smear produced by the supercoiled plasmid template, facilitating troubleshooting. The circular template plasmid is eliminated by digesting with DpnI, or a similar restriction enzyme, that cuts the methylated plasmid leaving the unmethylated PCR product.
The plasmid is typically dephosphorylated for ligation and amplification methods. However, it is possible to avoid this requirement (see alternative below).
Designing the insert.
The blunt-ended insert needs to be phosphorylated. If you plan to anneal oligonucleotides or use IDT Gene Fragments, such as gBlocks™ Gene Fragments, as your insert, note that these are not synthesized with 5’ phosphate groups unless requested—make sure to select the 5’ phosphate option when ordering these sequences for blunt-end cloning. Not all IDT fragment products are available with 5’ phosphate groups. For these, phosphates can be added to the 5’ ends of dsDNA with a simple kinase reaction, for example using the commercially available T4 Polynucleotide Kinase. However, this method is less efficient, particularly when bases other than guanine are kinase targets.
When the ends of the insert are not blunt, a polishing or filling reaction is required. Examples of ends that need polishing or filling include inserts generated by shearing or sonication, or by Taq polymerase, which preferentially leaves a single adenosine overhang at the 3’ ends; inserts produced by restriction digests; and some inserts produced by annealing multiple oligonucleotides to create longer products. A number of DNA polymerases will remove DNA overhangs and/or can be used to fill in missing bases if there is a 3’ hydroxyl available for priming. Polymerases for such reactions include T4 DNA polymerase, PFU, and the Klenow Fragment of DNA polymerase I. There are many options and kits available so check with the manufacturers to determine which one works best for your application.
As with other cloning methods, shorter inserts will usually ligate more efficiently than longer ones. Manufacturers typically include size recommendations for plasmid inserts in the plasmid documentation as a useful guide when planning your cloning experiment.
Finally, unlike cohesive end cloning, blunt-end cloning does not automatically recreate a restriction site following ligation of the insert unless bases are added to the insert sequence to complete the missing site (Figure 1).
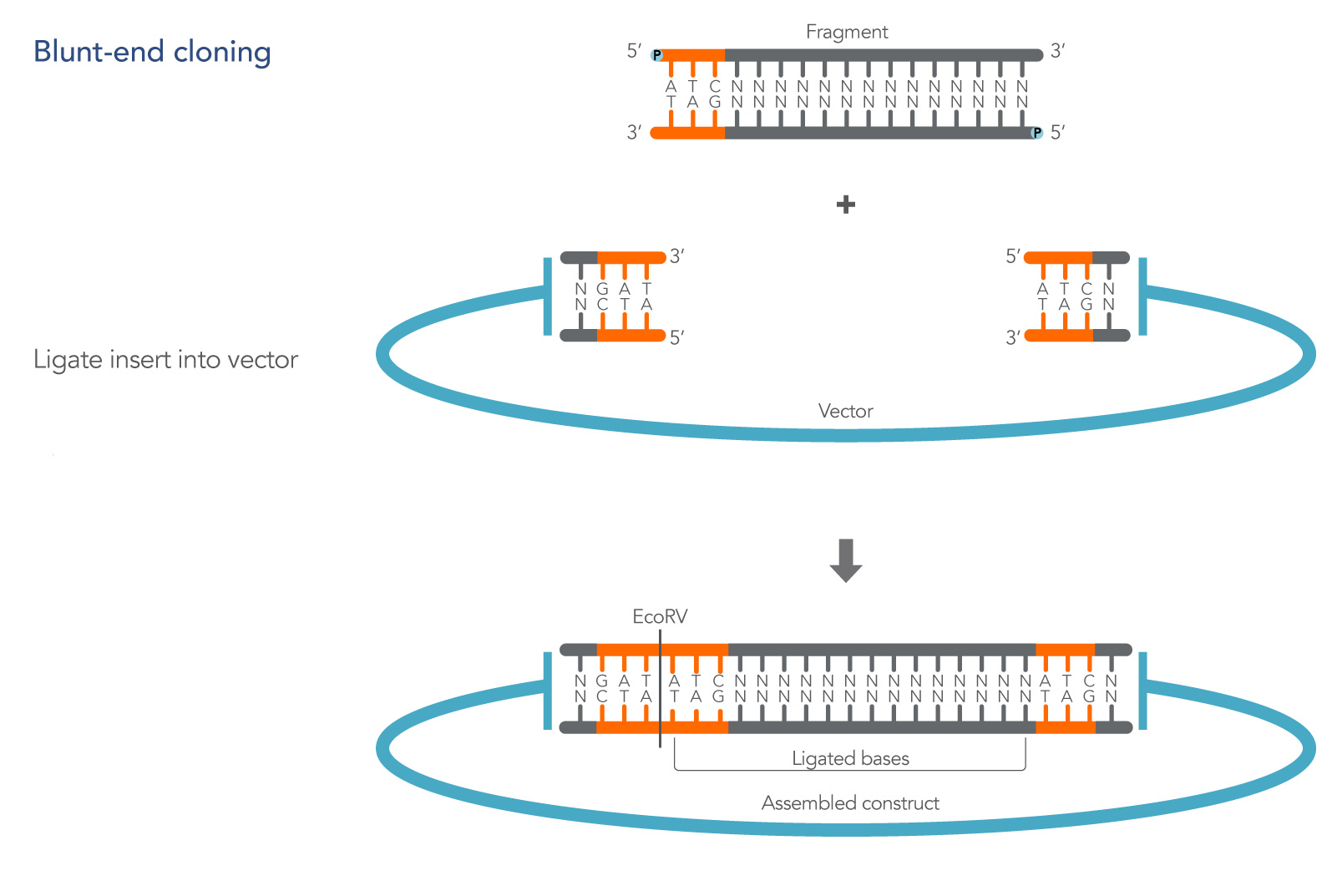
3'-OH and 5'-PO4 group, preventing the possibility of re-ligation and the formation of an empty construct with no DNA fragment inserted. A newly single EcoRV site is created on one end of the assembled construct so that a possible re-cut can occur, as shown in the figure.
Ligation conditions.
In blunt-end ligations, the association of 5’ phosphate groups and 3’ hydroxyl groups is more transient than in cohesive end ligations. Because they lack the hydrogen bond stabilization of cohesive ends, blunt-end ligations are more sensitive to reaction conditions, especially to the concentrations of the reaction components.
The likelihood of an insert associating with a linearized plasmid is increased by having a high concentration of available insert blunt ends. However, the intramolecular circularization of the plasmid, after one end of the insert has been joined, works best at lower concentrations. Insert concentrations that are too high, or overall DNA concentrations that are too high, can result in plasmids with multiple inserts, or concatemers. Although it is not always necessary, some researchers perform a short 1 hour incubation with high concentrations of insert and ligase, then dilute the reaction 20X with ligase buffer and allow the ligation to proceed for 4 more hours to facilitate the second step in the ligation reaction [1].
T4 ligase quality and concentration are also important. Blunt-end ligations typically take place in the presence of higher concentrations of ligase than cohesive end ligations. For example, whereas a cohesive end ligation may use
1-unit T4 ligase/20 μL reaction, a blunt reaction may use up to 3 units/20 μL reaction. Commercially available T4 ligases typically state whether they are optimized for blunt-end ligations or not. Follow the manufacturer’s guidelines to determine which ligase is appropriate for your cloning experiment and what concentration to use. Note that Taq ligase as well as several other ligases do not ligate blunt ends.
An alternative blunt-end method
As was mentioned, unless the insert is designed with the necessary bases to recreate the restriction site, the blunt restriction site used to linearize the vector is not normally recreated by the ligation of the insert (Figure 1). This allows for an alternative, less common, blunt-end cloning method that does not require the vector to be dephosphorylated. Instead, it relies on competing digestion and ligation reactions to decrease empty vector background.
In this blunt-end cloning method, the circularized plasmid and insert are placed in a reaction mixture containing the blunt end producing restriction enzyme, as well as the T4 ligase. The circular plasmid is cut, and the insert ligated in a single tube reaction. Empty plasmid that is produced by T4 ligation is subsequently re-cut by the restriction enzyme. As long as the insert is not designed to produce that specific restriction site, all circularized plasmids should contain the desired insert.
Specifics for this method—also described with a polishing enzyme component—can be found in Sambrook and Russell [1]. One reason why researchers may want to avoid this technique is that it involves mixing multiple optimized enzyme buffers, which may adversely affect the activity of individual enzymes. Check with the enzyme manufacturer(s) to determine if your chosen enzymes are compatible with this method.
For additional information about cloning methods, download the IDT DNA Cloning Guide.
