Dual Index UMI Adapters reduce index hopping and improve variant identification
Overview
To reduce sequencing costs, multiple libraries are often multiplexed using sample-specific DNA indexes and sequenced simultaneously in a single run. This may result in index misassignment, i.e., sequencing reads may be assigned to the wrong index and discarded from downstream bioinformatic analyses. To reduce index misassignments, IDT offers unique, dual-matched indexed adapters designed with unique molecular identifiers (UMIs).

Introduction
Next generation sequencing is a massively parallel sequencing technology that has revolutionized the biological sciences because it offers high throughput and speed for DNA and RNA sequencing. Since modern NGS platforms generate massive amounts of data, multiple libraries are often pooled—or multiplexed—and sequenced simultaneously in a single run to reduce the sequencing costs. To be able to identify each read post sequencing, all DNA fragments are tagged—or barcoded—using sample-specific DNA indexes during the library preparation step.
Regardless of the library preparation method, sample multiplexing sometimes results in index misassignment (cross-talk) due to various mechanisms like index hopping or barcode contamination. As a result, sequencing reads for a sample may be assigned to the wrong index and will be discarded from downstream bioinformatic analyses.
To minimize the risk of index hopping, it is usually recommended to carefully choose the multiplexing strategy, to use dual indexed adapters, and to remove all free adapters during library preparation. Unique, dual-matched sample indexes (UDIs) use distinct P5 and P7 index sequences for each sample (Figure 1). This minimizes the number of reads obtained from sequencing that must be removed from analyses due to barcode contamination [1,2].
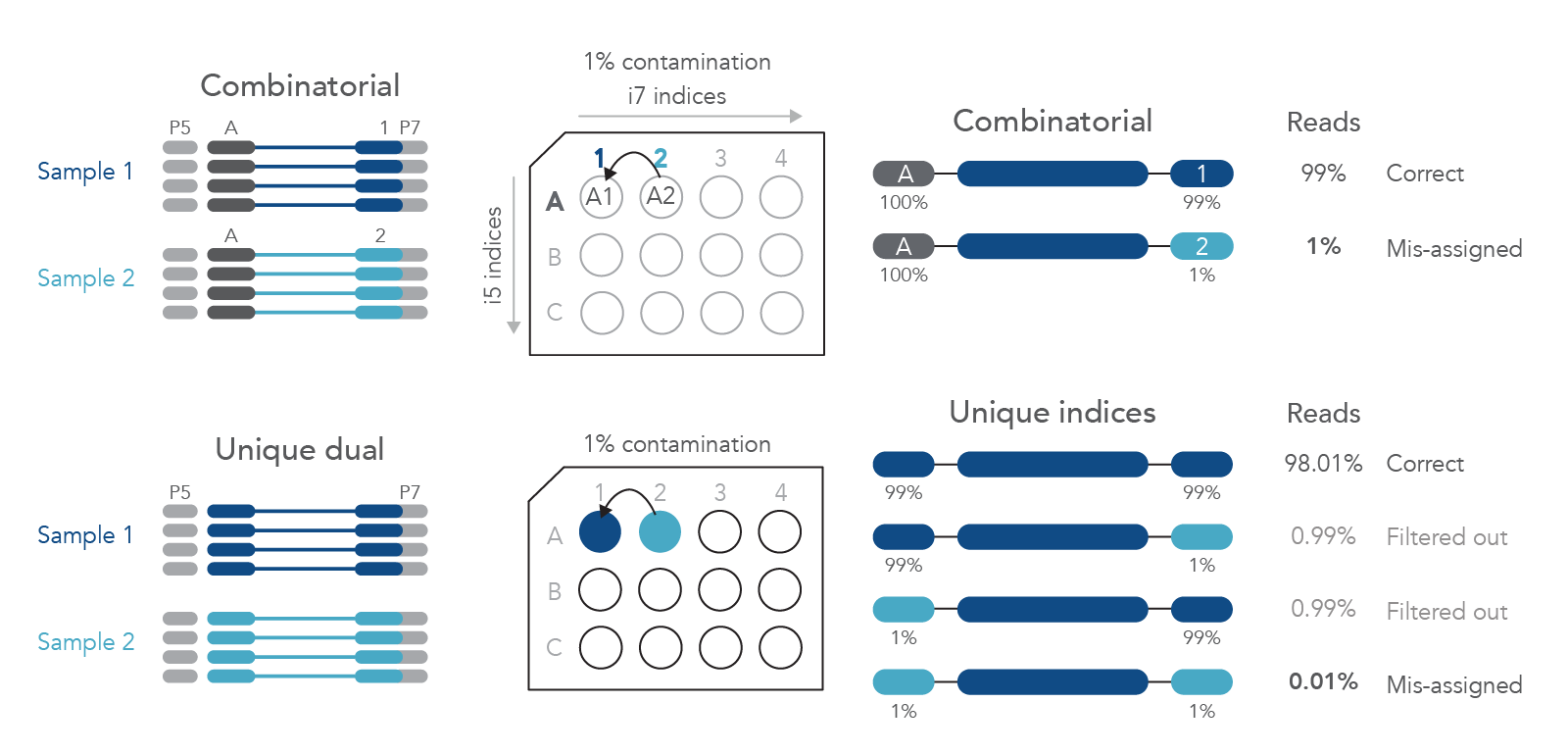
Figure 1. Unique, dual-matched sample indexes reduce read misassignment caused by barcode contamination. Example contamination of A1 adapter with 1% of A2 adapter. If only the i7 index is used to discriminate between the samples, 1% of sample A1 reads are misassigned to sample A2. If unique, dual-matched indexed adapters are used, only 0.01% of sample A1 reads are misassigned to sample A2 with the same 1% level of contamination.
However, in some cases, index hopping still occurs despite following all the recommended procedures. Higher levels of multiplexing lead to more pronounced cross-talk of library adapters which can be mediated through dual-index filtering during analysis (Figure 2).
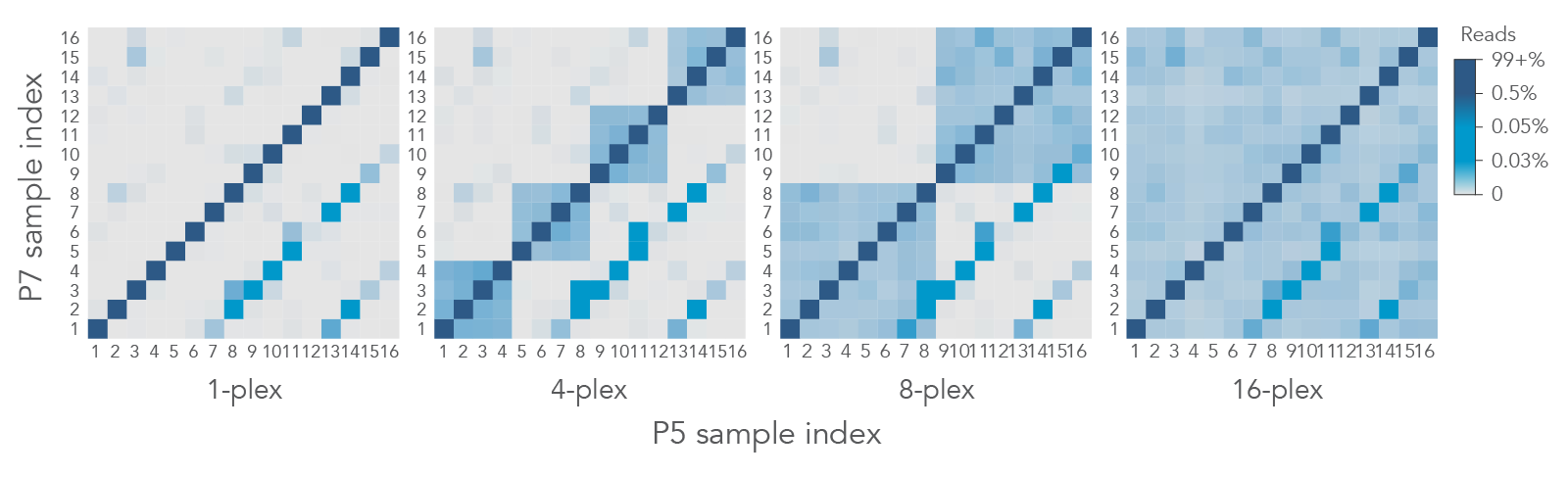
Figure 2. Unique, dual-matched indexes can identify contamination and index hopping events at different levels of multiplexing. Libraries were made from 250 ng of sheared genomic DNA (gDNA) and captured as 1-plex, 4-plex, 8-plex, or 16-plex captures using the xGen AML Cancer Panel (n = 1). Heatmaps show the percent of reads for all i5 and i7 sample index combinations in 1-, 4-, 8-, or 16-plex captures. The index hopping level of library adapters was 0.09% for single library captures but increased up to 0.39% for 16-plex captures.
To further reduce index misassignments, Integrated DNA Technologies, Inc. (IDT) offers unique, dual-matched indexed adapters designed with unique molecular identifiers (UMIs). These enable low-frequency variant identification in low-input and difficult-to-use samples. UMIs are short sequences that incorporate a unique barcode onto each individual molecule within a given sample library. These adapters are compatible with commercially available library construction kits and can be used for PCR-free applications. They can be analyzed as single-index (1), dual-index (2), or dual-index with UMI (3) (Figure 3).

Figure 3. Unique, dual-index adapters with UMIs can be read in one of three modes, depending on the resolution requirements of the experiment.
To assess the efficacy of the xGen™ UDI-UMI Adapters in resolving the issue of index hopping, well-characterized Genome in a Bottle (GIAB) cell lines and genotyped tumor-derived formalin-fixed and paraffin-embedded (FFPE) samples were sequenced from libraries constructed using the xGen UDI-UMI Adapters and enriched using a 75 kb custom xGen Custom Hyb Panel (Table 1). The use of unique, dual-matched indexed adapters reduced index cross-talk and improved the identification of rare variants (Figure 4 and Figure 5). Libraries constructed with xGen UDI-UMI Adapters provided uniform sequence coverage in cell-free (cf) and FFPE DNA samples (Table 2), further illustrating their application in variant identification (Figure 6 and 7).
Methods
Variant identification
Libraries with xGen UDI-UMI Adapters were prepared from 25 ng of the genomically well-characterized cell lines, NA12878/NA24385 (National Institute of General Medical Sciences (NIGMS) Human Genetic Cell Repository) to model low-frequency variants, as well as 25 ng, 50 ng, and 100 ng of genotyped tumor-derived FFPE samples. These libraries were enriched using a 75 kb custom xGen Custom Hyb Panel and sequenced on an Illumina® MiSeq™ v2 flow cell (Table 1). The same libraries were analyzed without the UMI data to represent the results from standard, combinatorial adapters. Variants were called in high GIAB confidence regions using VarDict [3] with start/stop deduplication or using a UMI consensus analysis.
Table 1. Details on sample mixtures used for assessing adapter performance with cell-line and FFPE DNA.
| Sample information | Cell-line | FFPE |
|---|---|---|
| Sample source | 99% NA12878 / 1% NA24835 | 99% Breast / 1% Stomach |
| Number of alternate SNPs | 10 (1%) / 44 (0.5%) | 20 (1%) / 56 (0.5%) |
| Library input | 25 ng | 25 / 50 / 100 ng |
| Total number of reads | ~18 million | 12 / 20 / 38 million |
Sequence coverage assessment
cfDNA samples collected from healthy donors (inputs of 5 ng, 10 ng, and 25 ng) and samples with FFPE DNA isolated from breast cancer tumor tissue (BioIVT®, inputs of 25 ng, 50 ng, and 100 ng) were used to generate libraries to determine the impact of xGen UDI-UMI Adapters on sequence coverage (Table 2). Following library construction, all samples underwent hybrid capture using a custom xGen™ Custom Hyb Panel spanning 75 kb.
Table 2. Details on samples used for assessing read coverage in libraries constructed with xGen Dual Index UMI Adapters
| Sample Information | Cell-free DNA | FFPE DNA |
|---|---|---|
| Sample source | Healthy Donors | Breast cancer tumor biopsy |
| Library input | 5 ng, 10 ng, 25 ng | 25 ng, 50 ng, 100 ng |
| Total Number of reads | 12 / 20 / 38 million | 20 / 40 / 60 million |
Results and discussion
The use of well-characterized human cell lines and previously genotyped tumor-derived samples helped to direct assess of how the xGen UDI-UMI Adapters addressed the issue of index hopping, a common problem resulting in the loss of reads when sequencing multiplexed libraries. To directly measure the efficiency of the xGen UDI-UMI Adapters, the same libraries were examined without the use of the UMI sequences.
In both the cell lines and tumor-derived FFPE samples, the use of UMI adapters improved positive predictive values (PPV) with a minimal impact on resolution (Figure 4A and Figure 5). UMI consensus calling also decreased the number of false-positive calls in the human cell lines from 136 to 4 (Figure 4B). In tumor-derived FFPE samples, consensus calling improved the PPV in all variants, even those with <1% allele frequency (Figure 5B).
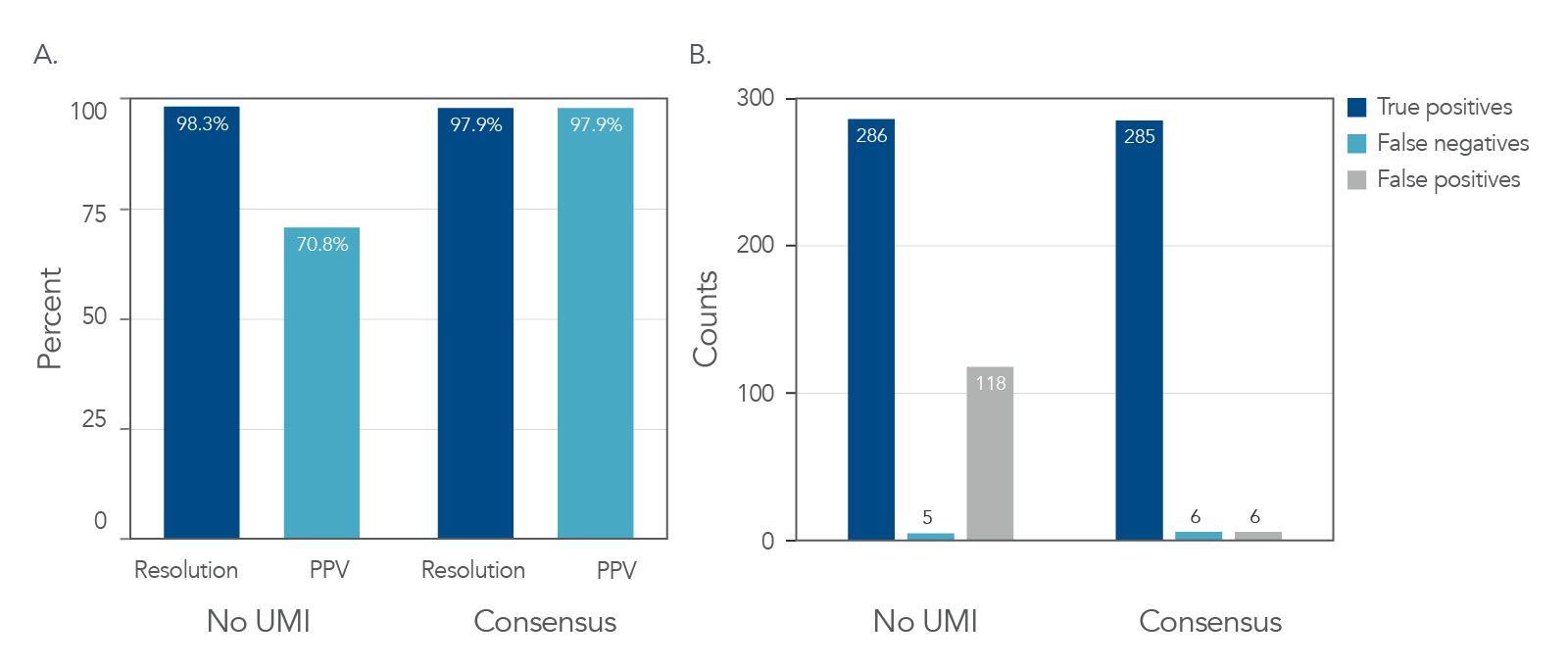
Figure 4. UMI consensus calling enhances variant calling while decreasing the number of false-positive calls in cell line samples. Libraries were generated using the xGen UDI-UMI Adapters and DNA collected from cell line samples. (A) UMI consensus calling improved PPV from 69.6% to 98.6% with minimal changes in resolution. (B) Total false-positive calls dropped from 136 to 4 with UMI consensus calling. (PPV = positive predictive value; PPV = True Positives/[True Positives + False Positives]).
Similarly, in tumor-derived FFPE samples, the xGen UDI-UMI Adapters increased PPV (Figure 5) with a minimal impact on resolution. More specifically, UMI adapters resulted in higher PPV in samples that contained variants with <1% allele frequency, meaning that these adapters increased the variant calling precision in these samples (Figure 5B).
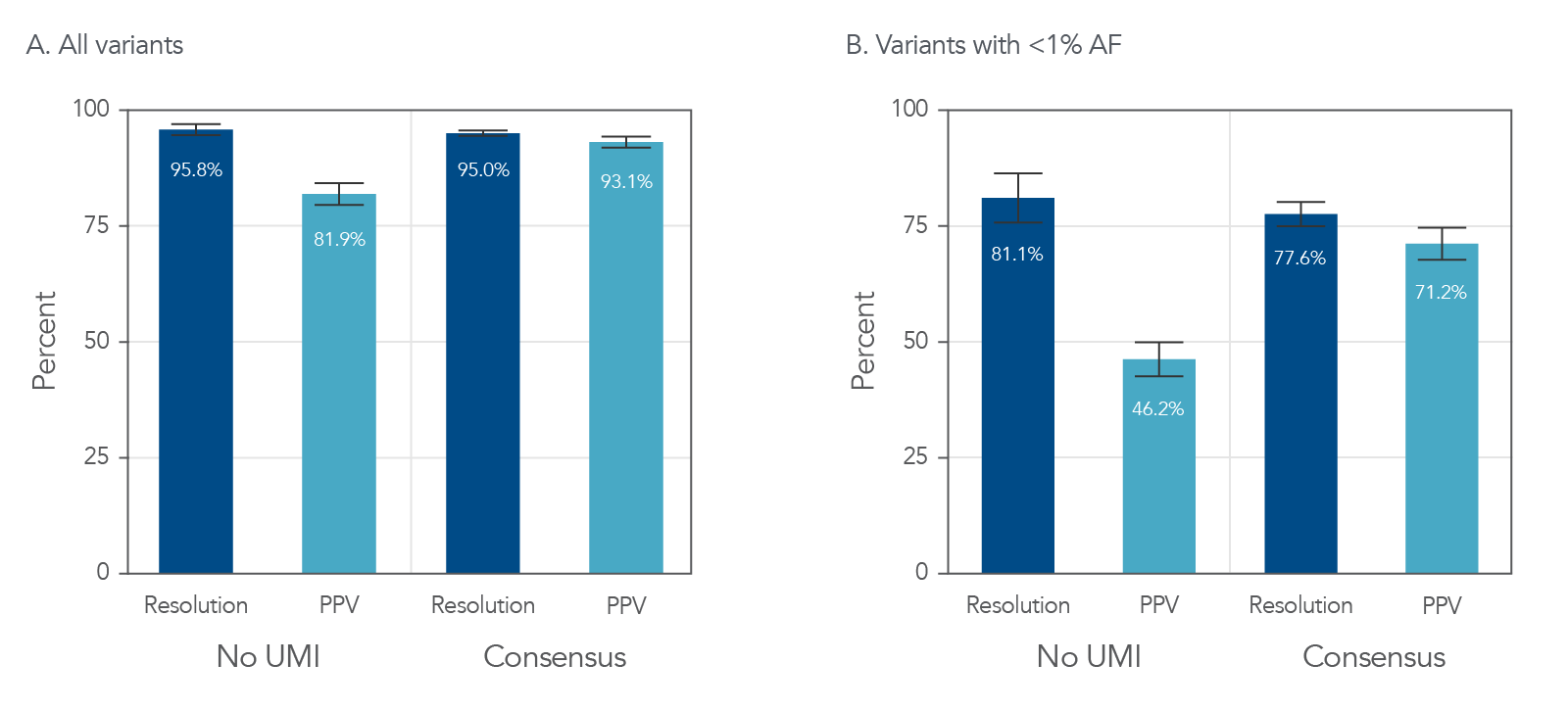
Figure 5. UMI consensus calling enhances variant calling in FFPE samples. Libraries were generated using the xGen UDI-UMI Adapters and DNA collected from tumor-derived FFPE samples. (A) Graph shows resolution and PPV using 0.6% min allele frequency for all mutations (n = 340). (B) Graph shows resolution and PPV using 0.6% min allele frequency for mutations present <1% (n = 76). (AF = allele frequency)
Another important factor in identifying variants is sequence coverage as uniform coverage results in precise variant identification without unnecessarily increasing costs of sequencing [1,4]. Therefore, xGen UDI-UMI Adapter containing libraries were generated from two sample types—tumor-derived FFPE DNA and cfDNA from healthy donors—and read coverage from each was evaluated. Libraries were generated from inputs ranging from 5–100 ng and hybrid captures were performed using an xGen Custom Hyb Panel spanning 75 kb. The percent of targets covered remained high at 100x, 300x and 500x sequence coverage, reflecting good uniformity of coverage (Figure 6 and Figure 7) for as low as 5 ng input and for both FFPE and cfDNA.
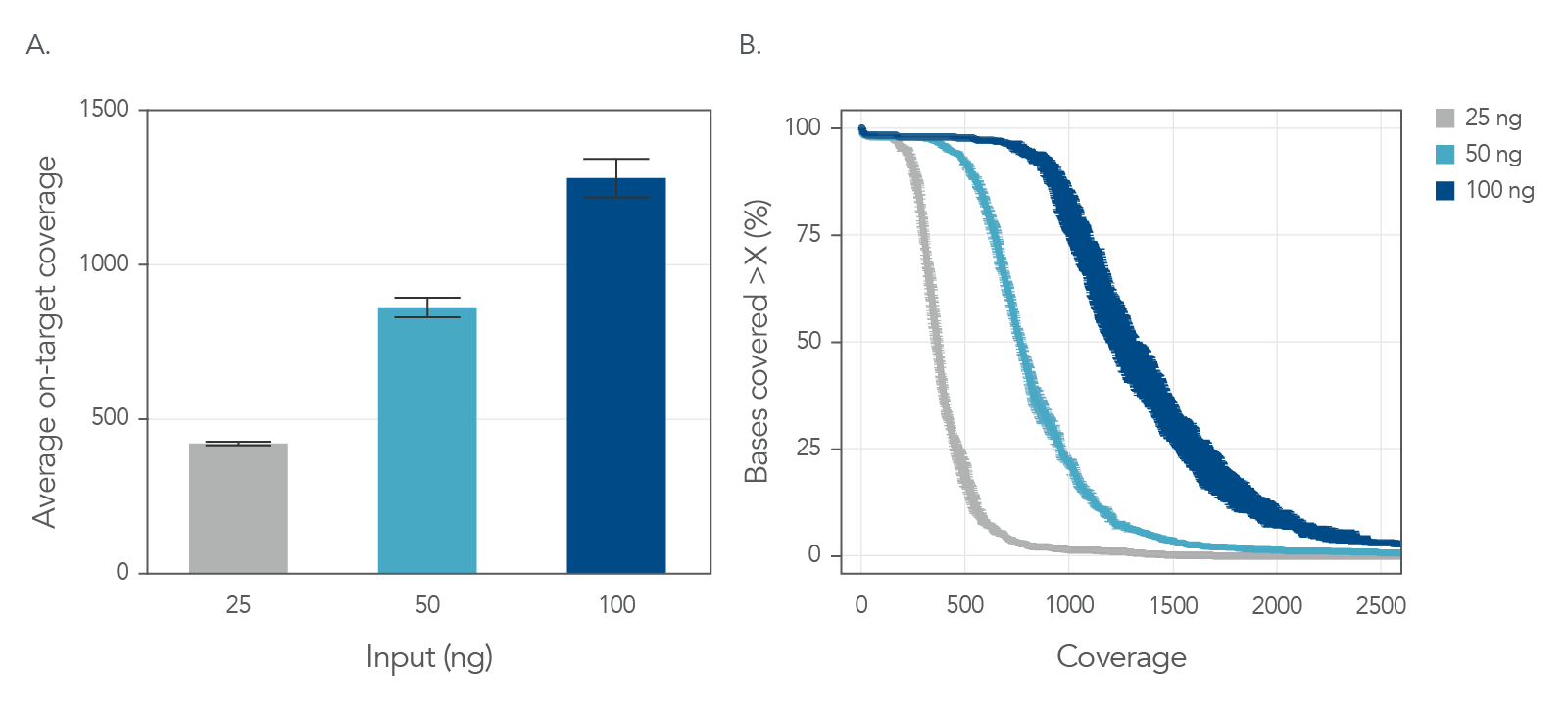
Figure 6. Uniform sequence coverage of FFPE DNA samples with various input material. Libraries were generated using the xGen UDI-UMI Adapters and DNA collected from tumor-derived FFPE samples. (A) Mean deduplicated coverage is shown for 25 ng, 50 ng, and 100 ng inputs using 20 M, 40 M, approximately 60 M total reads (56.9, 59.5, 60.6M), respectively. Error bars reflect standard deviation of replicates (n = 3). (B) Mean cumulative coverage by sample input is shown. Error bars on x-axis reflect standard deviation of coverage (n = 3).
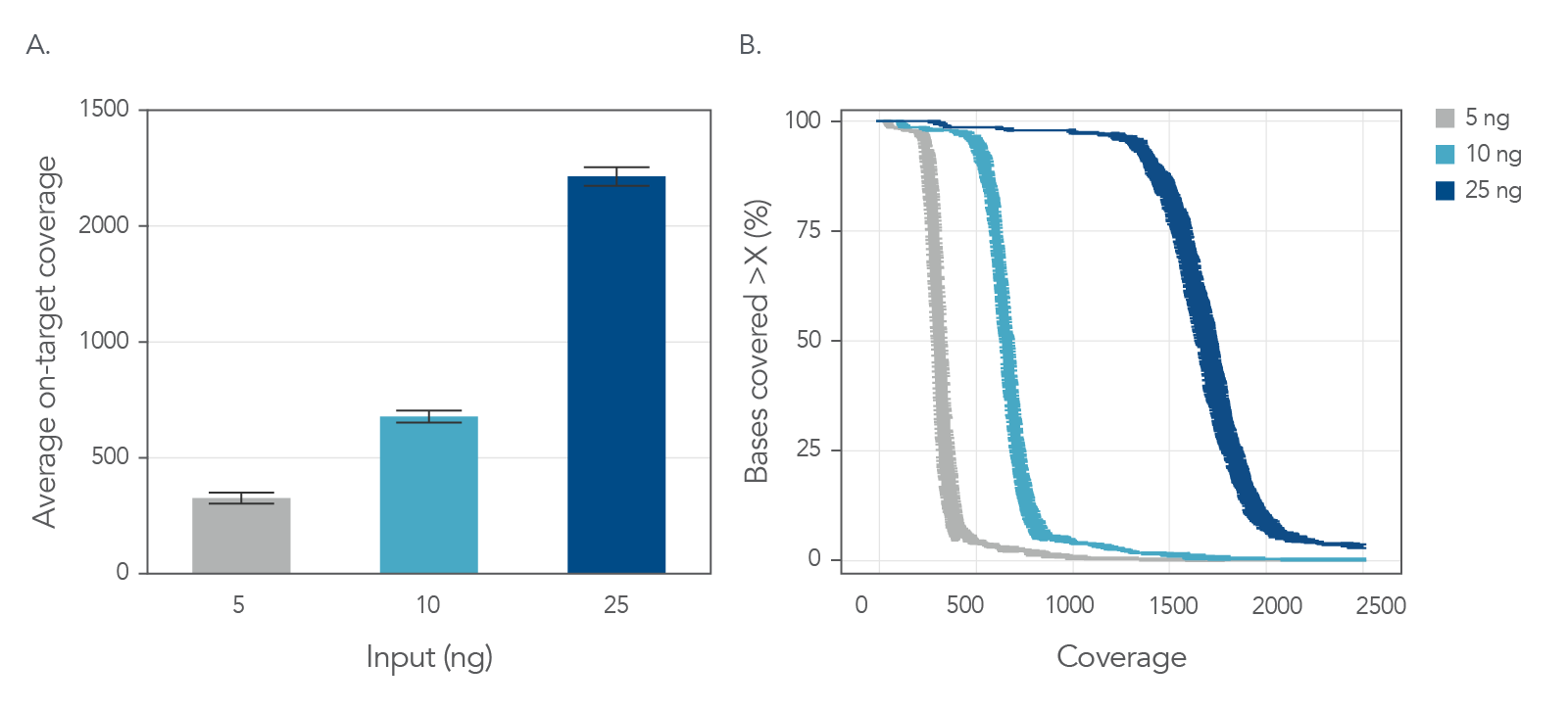
Figure 7. Uniform sequence coverage of cfDNA samples with various input material. Libraries were generated using the xGen UDI-UMI Adapters and tumor-derived cfDNA samples. (A) Mean deduplicated coverage is shown for 5 ng, 10 ng, and 25 ng inputs using 12 M, 20 M, and 38 M total reads, respectively. Error bars reflect standard deviation of replicates (n = 3). (B) Graph shows the mean cumulative coverage by sample input. Error bars on x-axis reflect standard deviation of coverage (n = 3).
Conclusions
Multiplexing of NGS samples can result in the loss of sequencing reads due to index cross-talk resulting from index hopping. The use of unique, dual-matched indexed (UDI) adapters can reduce read misassignment and increase the efficiency of NGS approaches in various research fields [2]. Combining UDI adapters with UMIs reduces information loss due to index hopping even further and enables more precise variant identification in low-input (5 ng) and difficult-to-use samples (cfDNA or FFPE), e.g., in cancer and SARS-CoV-2 research [5,6]. The results presented above show that the IDT xGen UDI-UMI Adapters increase the efficiency of variant identification and reduce the number of false-positive calls, leading to a more thorough and precise approach to distinguish rare variants in multiplexed studies. The use of xGen UDI-UMI Adapters results in uniform sequence coverage even in difficult-to-use samples demonstrating that these adapters can be used to identify variants in high-resolution studies without driving up sequencing costs.
Find more information on xGen UDI-UMI Adapters. Or read about how researchers are using xGen Dual Index UMI Adapters in the references [1,5,6] below.
References
- MacConaill LE, Burns RT, Nag A, et al. Unique, dual-indexed sequencing adapters with UMIs effectively eliminate index cross-talk and significantly improve sensitivity of massively parallel sequencing. BMC Genomics. 2018;19(1):30.
- Kircher M, Sawyer S, Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012;40(1):e3.
- Lai Z, Markovets A, Ahdesmaki M, et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016;44(11):e108.
- Sims D, Sudbery I, Ilott NE, et al. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. 2014;15(2):121-132.
- Alcoceba M, Garcia-Alvarez M, Chillon MC, et al. Liquid biopsy: a non-invasive approach for Hodgkin lymphoma genotyping. Br J Haematol. 2021;195(4):542-551.
- Wong CH, Ngan CY, Goldfeder RL, et al. Reduced subgenomic RNA expression is a molecular indicator of asymptomatic SARS-CoV-2 infection. Commun Med (Lond). 2021;1:33.