xGen™ Custom Amplicon Panels
Targeted sequencing of your most important genes
Tailored to meet your research needs, custom amplicon panels for Illumina ™ and Oxford nanopore technologies ™ offer a completely curated targeted sequencing workflow to interrogate genomic targets relevant to you.
xGen NGS—made to discover.
Ordering
- Multiplex hundreds to thousands of targets in a single tube PCR
- Compatible with low-input samples using 2.5-hour workflow
- xGen Normalase™ compatible: Supports high-throughput library quantification in a single enzymatic reaction
- Designed for researching germline and somatic variants, or targeted pathogen sequencing
- Provides high on-target and coverage uniformity
- Support available to help design your panel for Illumina or ONT sequencers
xGen Custom Amplicon Panels
This technology enables design of custom panels for targeted sequencing, offering a completely curated, targeted NGS workflow to rapidly interrogate genomic targets relevant to your research. xGen Amplicon technology’s ability to generate super amplicons result in increased coverage, even in changing or diverse genomes. Optional xGen™ Normalase module included to support high-throughput library quantification in a single enzymatic reaction.
Core Kit and Indexing sold separately.
Create designs for custom amplicon panels
To submit a design request, use the form and template. All custom panels are formatted in 384 rxn. Customers can choose a pool and ship option or a pool, and ship option with functional NGS testing and panel optimization.
Order my design
Upload your predesigned sequences by logging into your IDT customer account and filling out our order form. All custom panels are formatted in 384 rxn.
Any xGen Custom Amplicon Panel and Indexes for Oxford Nanopore Technologies™ will require a custom part number to be generated at point of order.
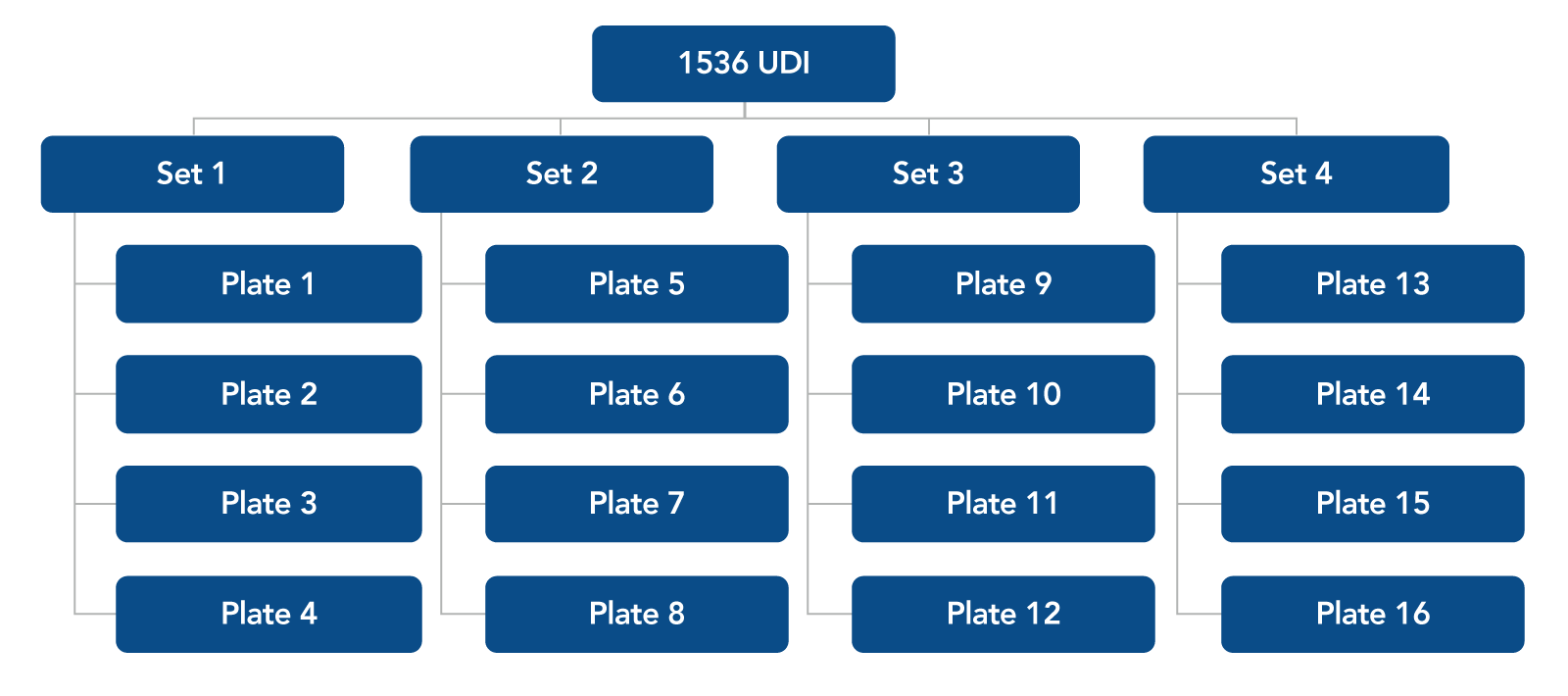
Figure 1. Plate combinations for 1536 UDI primer pairs. Each 96-well plate is sold in a set of four plates. For 1536 UDI primers, order Set 1, 2, 3, and 4.
For plate layouts of each of the 16 UDI plates offered see the xGen™ Normalase™ Indexing Primers quick reference guide.
xGen Custom Amplicon HS Panel
This technology enables low-frequency variant identification ≤1% through the incorporation of molecular identifiers (MIDs or UMIs), to reduce the rate of false-positive variant calls and increase variant identification introduced during PCR and sequencing.
Create designs for custom amplicon panels
To submit a design request, use the form and template. All custom panels are formatted in 384 rxn. Customers can choose a pool and ship option or a pool, and ship option with functional NGS testing and panel optimization.
Order my design
Upload your predesigned sequences by logging into your IDT customer account and filling out our order form. All custom panels are formatted in 384 rxn.
Any xGen Custom Amplicon Panel (HS included) will require a custom part number to be generated at point of order.
For customers who wish to purchase the legacy rhAmpSeq™, please contact us today.
Request a consultation
Want to learn more about our xGen™ Amplicon Panels—developed with super amplicon technology—and how to customize your panel or spike-in genes in our predesigned panels? Your time is valuable—we’ll prioritize your inquiry and be in touch to discuss it ASAP.
Request a consultationProduct details
xGen Custom Amplicon Panels are custom-designed primer pools to create multiple overlapping amplicons for specific genetic targets in research studies (Figure 1). Panels can multiplex hundreds to thousands of targets in a single tube
using a simple 2.5-hour workflow (Figure 2). Compatible with cell-free DNA and FFPE (formalin-fixed paraffin embedded) research samples starting with as little as 10 ng input or pathogen RNA/DNA in the picogram range. For a listing of the specifications
for custom panels, see Table 1. If multiple libraries are being examined at the same time, the final NGS library can be normalized using the IDT proprietary xGen Normalase reagent.
The cost-effective and flexible custom design means the
panel can be used for research applications such as:
- Genotyping by sequencing
- Variant identification with a limit of detection down to 1%
- Identifying germline inherited SNPs and indels
- Somatic cell variant identification
- Targeted virus or pathogen sequencing
- Genetic fingerprinting
- Liquid biopsy
Transform Your NGS Workflow with Automation
Looking to streamline your NGS workflows? Discover how automation can enhance efficiency and consistency in your lab with our NGS Automation solutions.
xGen Custom Amplicon Panels
EFFICIENT NGS THROUGHPUT & SCALABILITY
SIMPLEST, FASTEST WORKFLOW AVAILABLE
TAILORED GENOMIC CONTENT FOR LIBRARY PREP
xGen Custom Amplicon Panels are used to create targeted NGS libraries for research studies. Whether you need to examine hundreds or thousands of targets, the xGen Custom Amplicon Panels can be tailored to your specific research needs. Once created, the panel is used in a simple, one-tube workflow that is compatible with high-throughput applications.
Table 1. Product specifications of the xGen Custom Amplicon Panels
| Feature | Specification |
|---|---|
| Input DNA range | 10–25 ng of amplifiable DNA |
| Amplicon size | Versatile size for compatibility with many research sample types: whole blood, cell culture, FFPE, cfDNA (default maximum is 150 bp) |
| Custom design coverage | >90% of requested bases |
| Workflow time | Single-tube with 2.5-hour DNA-to-library workflow |
| Components provided | Custom primer pool |
| Limit of detection | As low as 1% allele frequency for variants |
| Panel size range | 15–1500 amplicons per panel; other options available upon request |
| Library multiplexing capability | Depends on type of indexing primers used |
| On-target % | >90% on-target reads |
| Coverage uniformity | >80% coverage uniformity at a >0.2 of mean depth |
| Compatible platforms | Illumina® Oxford Nanopore Technologies™ |
Simple, 2.5-hour workflow
From DNA to NGS library in as little as 2.5 hours, the workflow for the xGen Custom Amplicon Panels can be done in a single tube with minimal hands-on steps. As shown in Figure 2, the DNA sample is first amplified with the multiplex custom panel to create copies of each of your intended targets. Then the amplicons are converted into indexed libraries in a second PCR reaction with the appropriate indexing primers for your NGS instrument. When multiple libraries are pooled, IDT-proprietary xGen Normalase reagent is used to normalize the libraries for NGS analysis.
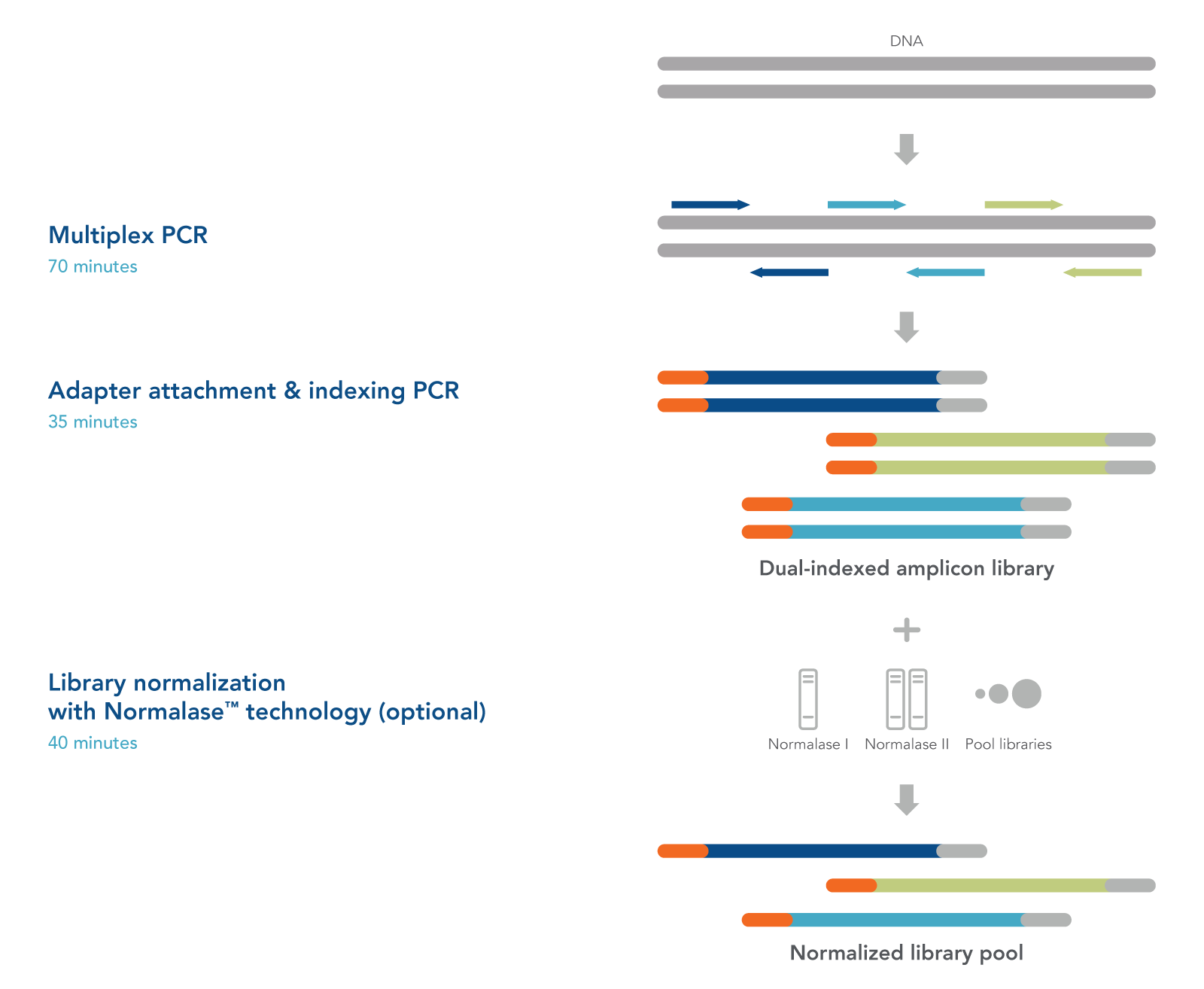
Figure 2. xGen Custom Amplicon Panels have a single tube workflow that is done in as little as 2.5 hours. Creating an NGS library starts with multiplex PCR. Your custom panel is combined with the DNA sample to amplify the targets of interest. The samples are then amplified with indexing primers to create a functional dual indexed library. As an optional step, the xGen Normalase reagent can be used after pooling multiple libraries to ensure each is equally represented in the final sample for the flowcell.
Resources
Frequently asked questions
Which sample types are compatible with xGen™ Custom Amplicon Panels?
Our xGen Custom Amplicon Panels are compatible with (but not limited to) the following sample types:
- Extracted genomic DNA (gDNA)
- Formalin-fixed, paraffin-embedded (FFPE) DNA
- Cell-free DNA (cfDNA)
- Viral RNA samples
- Viral DNA samples
Why do my xGen™ Amplicon libraries have low yields and contain primer dimers?
Low library yields and primer dimers may indicate one or more of the following:
- Using too low of an input DNA amount, or using damaged DNA that is poorly amplified
- Not setting up the multiplex PCR master mix and reactions on ice
- Not pre-setting your thermocycler program to temperature before adding samples (letting the thermocycler reach reaction starting temperature with samples in the block)
- Insufficient SPRI clean-ups
How do I analyze xGen™ HS EGFR Pathway Amplicon Panel data?
There are a few key considerations when analyzing sequencing data generated from the xGen HS EGFR Pathway Amplicon Panel with unique molecular identifiers (UMIs).
The first 10 bases in front of Read 2 constitute a UMI. For these first ten bases we recommend trimming them with Trimmomatic (using the CROP option) to make an MID/UMI fastq file for use with the MID pipeline from the fgbio package (Fulcrum Genomics). Before aligning the reads, make sure that the 10 bp UMI (which contains random bases) has been trimmed from 5’ of Read 2.
Also, check that adapter trimming is enabled while setting up the sequencing run. Alternatively, adapter trimming can be performed bioinformatically before analysis.
xGen Custom Amplicon Panels are designed with overlapping amplicons to provide contiguous regions of coverage in a single-tube format. Synthetic primer sequences will be encountered both at the beginning and end of some reads which must be trimmed during analysis. This can be done using a publicly available tool called Primerclip. See our app note Primerclip—A Tool for Trimming Primer Sequences for detailed information.
For more advice, reach out to our Scientific Application Supports team.
Note: A target BED file is provided with purchase of the xGen HS EGFR Pathway Amplicon Panel or the xGen Custom Amplicon Panel.
Can I use my own UDI primers in the xGen™ Amplicon Sequencing workflow?
Yes.
Please contact Scientific Application Support team if you would like assistance confirming compatibility of your own primers with the xGen Amplicon Sequencing workflow.
Note that using your own UDI primers without Normalase™ modifications will make the amplicon libraries incompatible with the downstream Normalase workflow.
Can I order my own custom indexing primers (e.g., UDI) with appropriate modifications to function with Normalase™ technology in the xGen™ Amplicon workflow?
Yes.
Contact your local sales representative, distributor, or our Scientific Application Support team if you would like assistance in ordering custom Normalase™ indexing primers for the xGen Amplicon workflow.